被卡住的分子马达可能在渐冻症的发展中起作用
伊利诺斯大学芝加哥医学院的研究人员称,神经细胞内营养物质、蛋白质和信号分子的运输和输送减慢可能是神经退行性疾病渐冻症(ALS)发展的原因之一。
研究人员展示了一种与遗传性肌萎缩性侧索硬化症有关的基因突变是如何导致这些重要分子沿着神经元长轴突运输的延迟。
他们的研究结果发表在在线杂志上《公共科学图书馆•综合》6月12日。
运动神经元是人体最长的细胞之一,有些细胞可以延伸到人的一半高度,多达三英尺。如果所有的细胞积木都是在细胞核所在的细胞一端制造的,而在细胞的另一端却需要它们,这就产生了一个问题。
神经元在分子上相当于高速公路和运货车- - - - - -神经纤维还有运动蛋白——沿着它们的长轴突运动,来回运送物质。但当运输受阻,产品无法到达所需地点时,细胞就不能最佳地发挥作用。这些运输问题会导致神经元与其他神经元和肌肉失去联系。
“如果运输过程被延迟或减慢,细胞的末端就会耗尽所需的物质,并失去与邻近神经元的突触连接,”UIC解剖学和细胞生物学助理教授、该研究的共同首席研究员Gerardo Morfini说。“没有这些连接,细胞就会死亡。”
“细胞死亡是肌萎缩性侧索硬化症漫长疾病过程的最后阶段,”斯科特·布雷迪说,他是UIC教授和解剖学和医学系主任细胞生物学首席研究员。“我们想要了解病理过程导致细胞死亡的神经元"
神经科学家们知道,一种名为SOD1的蛋白质突变导致了10%的ALS遗传病例。90%的肌萎缩性侧索硬化症病例没有已知的病因,被称为零星病例。
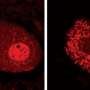
布雷迪和他的同事们之前曾用乌贼轴突的高分辨率视频显微镜证明,这种蛋白质的突变变体显著减缓了物质从细胞一端到另一端的运输。
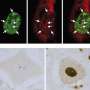
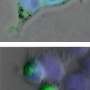
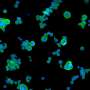
在新的研究中,研究人员观察了SOD1的突变形式是如何导致细胞运输放缓的。他们发现,这种突变的蛋白质激活了一种叫做p38激酶的分子,这种激酶反过来又修饰了一种主要的运动蛋白,它与神经轴突上的货物运输有关。这些修改马达蛋白与暴露于未突变SOD1的对照组相比,移动得很差。
他们还表明,通过同样的机制,突变的SOD1也减缓了转基因小鼠体内的转运。
“SOD1和p38激酶之间的通路可以为治疗肌萎缩性侧索硬化症的治疗干预提供有趣的靶点,无论是对某些遗传形式还是自发形式,SOD1的故障也是一个促成因素,”Brady说。