2016年11月21日报告
在基因医学的美丽新世界中找到你的诊断

(欧宝娱乐地址医学快报)-我们最近写了很多文章,关于一些了不起的人针对他们目前无法治愈的疾病开发个性化治疗方法。无论是罕见的孤儿病之类礼宾部主管综合症而且Castleman病这些患者能够利用自己的医疗专业知识,或其他经济和社会资源,创建资金充足的基金会和登记处,为他们的事业筹集支持。
然而,还有无数其他患者,无论年龄还是职业,他们都没有任何现成的人脉来获取内部知识和主动性。在很多情况下,特别是孤儿病,这些人甚至没有诊断他们可以用来为自己辩护。通常情况下,他们所拥有的只是一堆分散的论文报告和原始医学文献的复印出版物,没有明确的解释来将所有内容联系在一起。
更糟糕的是,它们所包含的复杂的遗传学细节,即使是专家也不一定能立即弄清楚。我现在知道这一点了(我不是医学专家或专业人士),因为我的一个邻居给了我一套这样的报告,我向一些商界精英寻求了明确的解释。目前的事态并不是任何人的直接过错,而是不完整和不断发展的知识体系的副作用,这种知识体系在表述上必然包含严重的模糊性。
下面我想介绍杰克逊·祖伯病例的重要基因结果(至少我试图理解它们),这是他的母亲艾米丽给我的。虽然显然不打算成为遗传学的完整入门,但应该有足够的细节,以便我们自己,以及任何专业的遗传学家、蛋白质实验家或建模师、神经学家、神经生物学家或放射学家临床医生可以提取更完整的图像,并有希望产生一些额外的探究线索。
杰克逊是遗传学家指定的“先证者”,意思是发起这项研究的人,在这种情况下是一个一岁的男孩。外显子组测序揭示了四个具有重大临床意义的基因的变异:
- NEB(星云素)c.11450G>A;p.S3817N
- PLP1(蛋白脂蛋白1)c.194T>G;p.I65S
- ERCC6(切除修复交叉互补组1)c.2924G>A;p.R975Q
- PGAP1 (gpi后附着蛋白1)c.2525+4C>T
医生们从逻辑上关注PLP1基因(并最初诊断出相关的Pelizaeus-Merzbacher病或“PMD”),因为它是一个x连锁纯合基因。这意味着杰克逊只有一个基因副本,并且特别容易受到该基因中任何有害突变的影响。其他三个基因位于“常染色体”上,杂合,因此不会立即成为主要的怀疑对象,因为存在另一个功能正常的基因副本。
这并不是说其他基因可以完全不考虑,特别是考虑到缺乏一个完整的基因组序列,它将包含任何潜在的外显子组序列中未分析的区域,包括通常在基因开始的调控区域。也有可能是一个基因拷贝根本不能提供所需的蛋白质阈值水平,或者是有缺陷的蛋白质本身导致了一些新的病理。
似乎所有基因的亲代起源都被确定了(所谓的“阶段”分析),但我不知道这种分析是否确定了假定的“良好”基因副本是否也可能具有相同的(因此未被检测到的)变体。
从先前的许多动物研究中,我们已经对NEB基因和蛋白质有了相当多的了解,尽管它通常在大脑中表达(除了相当关键的肌肉区域),但动物研究的结果表明,在有残疾的星云蛋白基因存在的情况下,基本上是正常的认知功能和神经结构。
PGAP1在技术上是杂合的,只有当还存在某种未被检测到的“复合杂合”突变(第二种坏变体或多态性)时,才会立即成为一个危险信号;换句话说,来自另一个假定健康的父母的基因有不同的突变,在这种情况下,严重的神经问题是已知的可能影响。PGAP1是产生GPI(糖基磷脂酰肌醇)所必需的,GPI附着在一些蛋白质上,髓鞘化少突胶质细胞将大量这些GPI蛋白质定向到髓鞘。
ERCC6参与转录偶联修复,类似地,只有当两个基因都受到影响时,ERCC6才通常与严重的神经疾病相关(如Cockayne综合征)。我只想在这里指出它在一些小头症结局中的意义,包括小头症和ATR (Seckle综合征)。如果有任何重要的问题出现在这里,一个测试错误的ERCC6修复能力可能是皮肤成纤维细胞的辐射敏感性,尽管我不知道它的准确性和信息。
为了进一步研究Jackson的PLP1变体的具体情况,我们首先需要解码并消除该变体的遗传标记;“c.194T > G;p.I65S”。我需要验证我在这里写的东西,因为很容易出错。
首字母c。’表明我们正在研究的是互补DNA,就像我们正在处理的那样外显子组测序信息。它是指以DNA (GCAT)碱基而不是RNA (GCAU)碱基表达的mRNA转录本序列。有一个“基因组测序”参考(g.)将在这里提供更多的信息,原因有很多,即存在多个转录起始位点(启动子),替代剪接,使用不同的poly-A添加信号,多个翻译起始位点(atg -密码子),以及长度变化的发生。潜在地,如果外显子组测序在mrna被编辑之后(无论是在核特异性或细胞质特异性编辑中),这也将是一个问题,尽管RNA编辑(转录后碱基的修改,主要是在人类中A到G或A到I的替换)相当罕见。
正如我所理解的符号194T>G,在Jackson的PLP1 cDNA中的第194个碱基对位置有一个G,而大多数正常的cDNA会有一个T。因为G(像a一样)是嘌呤,T(像C一样)是嘧啶,这种取代被称为“转位”,而不是“转换”(这将发生在嘌呤到嘌呤或嘧啶到嘧啶切换的情况下)。由于细胞中存在自然机制,可以更容易地将单环嘌呤转化为其他嘌呤,或将双环嘧啶转化为其他嘧啶,因此跃迁明显比转位更常见。
为了更好地了解这种T>G是如何产生的,我采访了谢菲尔德大学生物医学科学系教授、细胞生物学家卡尔·斯麦斯,以及贝尔法斯特健康与社会保健信托基金临床主任、遗传学家谢恩·麦基。“T>G”并不一定意味着基因中的G已经直接变成了T。例如,这种转换可以作为非编码链上从C到a的突变的结果出现。G-A碱基可以很好地配对(和其他一些碱基一样,尽管正常的配对是A到T和G到C),而不会在编码(感觉)和非编码(反义)链之间引起主要的结构问题。因此,在下一轮合成中,a会有一个T插入到相反的链中。
任何一条链都可能有原始的突变,DNA复制过程将为单个位点产生两个不同的编码序列。因此,一个人可以得到两个具有复杂表型的细胞。在非编码链C到a的突变后,得到稳定的G-A碱基对,复制后得到G-C对和T-A对,后者对应于Jackson的突变。这可能发生在减数分裂期间,也可能发生在精子中,也可能发生在有原始突变的人的发育过程中。
因为杰克逊从他的母亲那里继承了带有PLP1变体的X染色体,所以对他的祖父母进行了检查,发现他的祖父也有相同的变体。因为格兰普斯是无症状的,医生或多或少撤销了PMD的诊断。对这种情况的一种可能解释是,爷爷可能是一个“马赛克”。换句话说,这种突变不是在精子水平上出现的,而是在发育的后期出现的(作为一种体细胞突变),在这种情况下,产生格兰普斯神经系统的细胞可能有一个正常的PLP1副本,因此他非常正常。另一种可能性是,祖父自己继承了这种变异,但仍然能够在他的神经系统细胞中修复它。
虽然这是罕见的,也可以想象,杰克逊有相同的突变,但它是独立获得的,即。它又在杰克逊的血统中作为从头变体出现。也许当你想象无论格兰普斯突变起源于什么基因或代谢背景时,杰克逊也会出现类似的背景,这不是完全不可想象的。更典型的传统想法是,“自发”突变或多或少是在DNA合成等事件中随机出现的,因为在复制过程中存在一些不可忽略的错误率,从而逃脱了校对机制。
也有可能这种突变在格兰普斯的遗传背景中没有太大的影响,但在杰克逊的遗传背景中却有显著的影响,即。“促进性”突变对PMD是必要的,但不是充分的。PMD的一个奇怪特征是,高达70%的患者有重复的PLP1基因——一个额外的副本。杰克逊似乎明确地检查了这一点,因为外显子组测序没有发现它,但他没有复制。他的线粒体也被测序,发现是正常的,然而,面对并不罕见的线粒体异质性(不止一组独特的mtDNA),我们可能也很好奇这里实际上采样了什么线粒体来源。
一个重要的相关问题是,在外显子组分析中测序的组织来源是什么——是血液、皮肤还是上皮细胞?由于相同的基因在不同的组织中通常以不同的方式拼接,因此在不同组织的外显子组分析中也会给出不同的cdna。
为了克服这一隐现的诊断障碍,除了全序列分析之外,还可以进行功能蛋白研究,以尝试并确定变体PLP1替代的可能影响。这可以包括使用软件工具来模拟蛋白质的结构和功能,并在实验室中实际构建变异蛋白质并在动物身上表达以寻找效果。例如,通过创建所谓的条件敲入鼠标行,
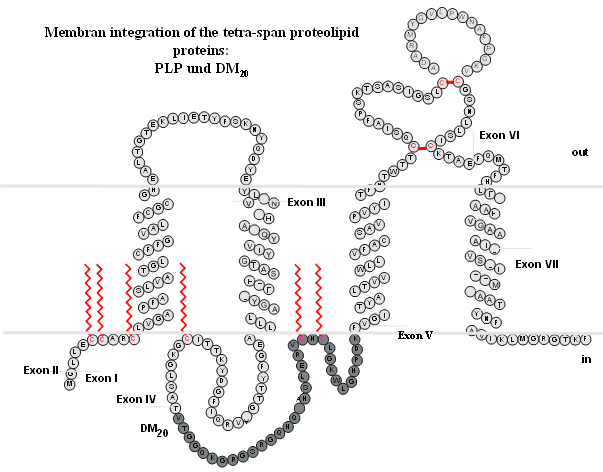
要开始这种蛋白质分析,我们需要看看变体符号的第二部分——“p.I65S”。这里的“p”表示我们正在谈论的是与cDNA或mRNA序列对应的蛋白质的氨基酸序列。它说Jackson的PLP1将在65号位置有一个丝氨酸(S)取代正常的异亮氨酸。
如果可能的异亮氨酸DNA密码子(意义)是ATT、ATC、ATA,而新的变体可能的丝氨酸密码子是TCT、TCC、TCA、TCG、AGT、AGC,我们可以通过消除过程假设是ATT或ATC苏氨酸的中间密码子点变成了AGT或AGC丝氨酸密码子。我认为,假设任何碱基对替换通常出于某种原因在细胞中产生和/或保持不修复是有道理的(即使这个原因是对皮肤细胞施加了过度的太阳辐射),而这个原因通常会反映出细胞的更大的背景代谢和环境,以及可能发生的生物体中正在发生的事情。
异亮氨酸是疏水氨基酸,丝氨酸是极性不带电氨基酸。这些是完全不同的动物,通常认为这种替代应该对蛋白质的结构或功能有重大影响。问题是效果如何?在检查这类事情的一些常用软件工具和数据库时,我们发现“PolyPhen2”说这种替换可能是有害的,“MutationTaster”也不满意,ExAC和1000G中都没有记录。
正常PLP1蛋白的一些不同剪接变体的典型膜结构在十多年前就已经确定了。这是一种高度保守的蛋白质,在从老鼠到人类的几个物种中几乎是相同的。最近,一些三维蛋白质构象,也就是实际的晶体结构,也被确定了,有时是与其他结合蛋白结合。假定的膜拓扑结构是4个跨膜螺旋,第65丝氨酸(或其位置取决于氨基酸开始计数的位置)位于第一个膜螺旋的胞外顶端。虽然丝氨酸可以在各种蛋白质中磷酸化,但不太可能在观察到的位置上磷酸化。
随着PLP基因的选择性剪接产生四种产物——经典的PLP和DM20蛋白脂,以及最近描述的srPLP和srDM20蛋白脂,尝试了解神经系统中各种细胞产生了多少这些不同的产物,以及它们对这些细胞的影响是很重要的。已经知道了很多,现在还在持续不断地获取更多的信息。此外,这些产物(亚细胞)定位到细胞内的各个隔室是一个重要的点(一些进入髓鞘,其他定位到线粒体,而其他留在内质网)。我认为,在每种情况下,主要的问题是,这种蛋白质是过多还是不足,以及在每种情况下,功能不良,无功能或其他阻碍蛋白质的影响是什么?到目前为止,我们已经知道,虽然过度表达PLP基因的转基因小鼠表现出神经元变性和轴突分裂,但可能矛盾的是,PLP缺失小鼠中PLP/DM20的缺失也会导致轴突肿胀。由于这种蛋白质通常非常丰富,约占髓鞘蛋白总量的50%,微小的变化会产生巨大的影响。
目前尚不清楚丝氨酸点是否会影响剪接(但请注意下图附近的剪接位点),或影响任何蛋白质的交联半胱氨酸,或影响任何关键的半胱氨酸棕榈酰化,但还需要进一步的研究。由于已知该蛋白质也形成二聚体,甚至可能形成更高的旧多聚体,可能相互连接,穿过致密的髓磷脂层,丝氨酸对这种寡聚的影响可能是一个重要的问题。尽管这些半胱氨酸大部分都更接近蛋白质的开始,但它们现在还是个谜,因为它们可以通过它们的硫基为蛋白质做很多事情。当交联或解联时,它们改变蛋白质构象,也转导氧化还原信号。当棕榈酰化时,它们瞄准并定位蛋白质到髓鞘,当以各种明确的定位序列或基序(如C-3xC或C-10X,如这里)间隔时,它们也以线粒体为目标,参与各种功能。
我认为,将杰克逊的详细资料分发给任何可能准备提供帮助的医生和研究人员是至关重要的,即各种“孤儿”进行性退行性神经疾病的专家。这类专家将包括各种影响髓磷脂的白质营养不良和溶酶体储存疾病,以及最终影响线粒体及其在能量产生和其他关键代谢过程中的作用的专家。我的脑海中立刻浮现出两个家伙,他们是圣路易斯华盛顿大学的布鲁诺·贝尼特斯(Bruno Benitez)和CHOP的道格·华莱士(Doug Wallace)。
我不会深入研究在基因测试中发现的其他变体,只是注意到PGAP1的符号与其他的略有不同,给出的是c.2525+4C>T。注释c.2525+4C>T似乎表明该变体位于距离最后一个外显子核苷酸+4个核苷酸的位置。这种变体被预测为“剪接供体”,这意味着它可以改变产生的蛋白质的长度,一种不同的转录本。PGAP1有22个外显子和至少11个剪接变体。该变异在内含子下游核苷酸位置2525处发生突变。这就造成了剪接结失败,内含子不会被剪接出来,因此变体将包含与内含子对应的蛋白质序列。
关于这是否会直接影响PGAP1的酶活性还有很多不确定性。它会生成一个蛋白质长度出乎意料的。受这种突变影响的剪接变异的精确数量需要仔细分析,与大脑相比,组织样本很可能具有不同的PGAP1变异谱。可能值得注意的是PGAP突变都与发育缺陷有关,当然,显然还需要更多的信息。
.
进一步探索
©2016 M欧宝娱乐地址edical Xpress